For details about building and using alphafold see alphafold 2.0.
Running alphafold 2.1.1 with docker
sample batch-script, and run script can be found in /software/alphafold/2.1.1. Singularity still needs to be done. To get started:
- Create a batch-script a sample is pasted below. Customize it to contain proper partitions and limits
- Note: --max_template_date=2020-05-1 is used to run casp14 tests for T1050. You probably don't want to use it in most cases!
- Use /software/alphafold/2.1.1/alphafold.sh or customize it according to your needs.
- For multimer: use AF_preset=multimer ..., the default is monomer.
- For multimer: each monomer has to be a separate entry with full sequence in the fasta-file, even if all monomers are identical
- Almost all parameter can be customized, see the table below for details
- sbatch <your-alphafold-script>
alphafold parameters:
| parameter | description | default | environment variable in alphafold.sh | default value of environment variable |
|---|---|---|---|---|
| --[no]benchmark | Run multiple JAX model evaluations to obtain a timing that excludes the compilation time, which should be more indicative of the time required for inferencing many proteins | false | none | none |
| --data_dir | Path to directory of supporting data | none | AF_datadir | /beegfs/desy/group/it/ReferenceData/alphafold |
| --bfd_database_path | Path to the BFD database for use by HHblits | none | AF_bfd | $AF_datadir/bfd/bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt |
| --db_preset | <full_dbs|reduced_dbs>: Choose preset MSA database configuration - smaller genetic database config (reduced_dbs) or full genetic databaseconfig (full_dbs) | full_dbs | none | none |
| --fasta_paths | Paths to FASTA files, each containing a prediction target that will be folded one after another. If a FASTA file contains multiple sequences, then it will be folded as a multimer. Paths should be separated by commas. All FASTA paths must have a unique basename as the basename is used to name the output directories for each prediction (a comma separated list) | none | none | none |
| --hhblits_binary_path | Path to the HHblits executable. | /usr/bin/hhblits | none | none |
| --hhsearch_binary_path | Path to the HHsearch executable. | /usr/bin/hhsearch | none | none |
| --hmmbuild_binary_path | Path to the hmmbuild executable. | /usr/bin/hmmbuild | none | none |
| --hmmsearch_binary_path | Path to the hmmsearch executable. | /usr/bin/hmmsearch | none | none |
| --is_prokaryote_list | Optional for multimer system, not used by the single chain system. This list should contain a boolean for each fasta specifying true where the target complex is from a prokaryote, and false where it is not, or where the origin is unknown. These values determine the pairing method for the MSA (a comma separated list) | none | none | none |
| --jackhmmer_binary_path | Path to the JackHMMER executable. | /usr/bin/jackhmmer | none | none |
| --kalign_binary_path | Path to the Kalign executable. | /usr/bin/kalign | none | none |
| --max_template_date | Maximum template release date to consider. Important if folding historical test sets. | none | none | none |
| --mgnify_database_path | Path to the MGnify database for use by JackHMMER | none | AF_mgnify | $AF_datadir/mgnify/mgy_clusters.fa |
| --model_preset | <monomer|monomer_casp14|monomer_ptm|multimer>: Choose preset model configuration - the monomer model, the monomer model with extra ensembling, monomer model with pTM head, or multimer model | monomer | AF_preset | monomer |
| --obsolete_pdbs_path | Path to file containing a mapping from obsolete PDB IDs to the PDB IDs of their replacements. | none | AF_obsolete | $AF_datadir/pdb_mmcif/obsolete.dat |
| --output_dir | Path to a directory that will store the results. | none | AF_outdir | /tmp/alphafold |
| --pdb70_database_path | Path to the PDB70 database for use by HHsearch. | none | AF_pdb70 | $AF_datadir/pdb70/pdb70 |
| --pdb_seqres_database_path | Path to the PDB seqres database for use by hmmsearch. | none | AF_pdbseqres | $AF_datadir/pdb_seqres/pdb_seqres.txt |
| --random_seed | The random seed for the data pipeline. By default, this is randomly generated. Note that even if this is set, Alphafold may still not be deterministic, because processes like GPU inference are nondeterministic (an integer) | none | none | none |
| --small_bfd_database_path | Path to the small version of BFD used with the "reduced_dbs" preset. | none | none | none |
| --template_mmcif_dir | Path to a directory with template mmCIF structures, each named <pdb_id>.cif | none | AF_mmcif | $AF_datadir/pdb_mmcif/mmcif_files |
| --uniclust30_database_path | Path to the Uniclust30 database for use by HHblits. | none | AF_uniclust30 | $AF_datadir/uniclust30/uniclust30_2018_08/uniclust30_2018_08 |
| --uniprot_database_path | Path to the Uniprot database for use by JackHMMer. | none | AF_uniprot | $AF_datadir/uniprot/uniprot.fasta |
| --uniref90_database_path | Path to the Uniref90 database for use by JackHMMER. | none | AF_uniref90 | $AF_datadir/uniref90/uniref90.fasta |
| --[no]use_precomputed_msas | Whether to read MSAs that have been written to disk. WARNING: This will not check if the sequence, database or configuration have changed. | false | none | none |
/software/alphafold/2.1.1/alphafold.sh predefines a few more custom variables which can be redefined on the command line of the batch script:
| environment variable | description | default value |
|---|---|---|
| AF_docker | docker runtime environment | --gpus all -u `id -u`:`id -g` --userns=host --security-opt no-new-privileges |
| AF_mount | volumes mounted in the docker container | -v /tmp:/tmp -v $HOME:$HOME -v /beegfs/desy:/beegfs/desy -v /beegfs/cssb:/beegfs/cssb -v /software:/software |
| AF_image | docker image | gitlab.desy.de:5555/frank.schluenzen/alphafold:2.1.1 |
| AF_env | GPU memory handling | --env TF_FORCE_UNIFIED_MEMORY=1 --env XLA_PYTHON_CLIENT_MEM_FRACTION=4.0 --env XLA_PYTHON_CLIENT_ALLOCATOR=platform |
| AF_models | Naming template | model_1,model_2,model_3,model_4,model_5 |
Databases can be found in /beegfs/desy/group/it/ReferenceData/alphafold/, but feel free to use your own set of DBs. small_bfd is not defined in the sample script, but can be found at /beegfs/desy/group/it/ReferenceData/alphafold/small_bfd/bfd-first_non_consensus_sequences.fasta. Last update: mid November 2021.
sample batch script
#!/bin/bash
#SBATCH --partition=maxgpu
#SBATCH --constraint='(A100|V100)&BS'
#SBATCH --time=0-12:00
#SBATCH --job-name=T1050
unset LD_PRELOAD
AF_outdir=/beegfs/desy/user/$USER/ALPHAFOLD2.1 /software/alphafold/2.1.1/alphafold.sh \
--fasta_paths=/software/alphafold/2.0/T1050.fasta --max_template_date=2020-05-1
# for a multimer:
AF_preset=multimer AF_outdir=/beegfs/desy/user/$USER/ALPHAFOLD2.1 /software/alphafold/2.1.1/alphafold.sh \
--fasta_paths=/software/alphafold/2.0/T1050-2.fasta --max_template_date=2020-05-1
exit
Note: the constraint has to contain BS to select nodes with sufficiently big scratch folder. It's a temporary workaround, otherwise docker will likely fail due to space limitations.
sample run script
#!/bin/bash
# basic setup
unset LD_PRELOAD
source /etc/profile.d/modules.sh
module purge
module load maxwell cuda/11.3
# alphafold basics
export AF_NVL=/usr/local/nvidia/lib:/usr/local/nvidia/lib64:/software/cuda/cuda-11.3/targets/x86_64-linux/lib:/software/cuda/cuda-11.3/lib64
export PATH=/software/alphafold/2.1.1/python3.6/bin:$PATH
export PYTHONPATH=/software/alphafold/2.1.1/python3.6:$PYTHONPATH
export AF_datadir=${AF_datadir:-/beegfs/desy/group/it/ReferenceData/alphafold}
# databases
AF_uniref90=${AF_uniref90:-$AF_datadir/uniref90/uniref90.fasta}
AF_bfd=${AF_bfd:-$AF_datadir/bfd/bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt}
AF_mmcif=${AF_mmcif:-$AF_datadir/pdb_mmcif/mmcif_files}
AF_obsolete=${AF_obsolete:-$AF_datadir/pdb_mmcif/obsolete.dat}
AF_pdb70=${AF_pdb70:-$AF_datadir/pdb70/pdb70}
AF_mgnify=${AF_mgnify:-$AF_datadir/mgnify/mgy_clusters.fa}
AF_uniclust30=${AF_uniclust30:-$AF_datadir/uniclust30/uniclust30_2018_08/uniclust30_2018_08}
AF_uniprot=${AF_uniprot:-$AF_datadir/uniprot/uniprot.fasta}
AF_pdbseqres=${AF_pdbseqres:-$AF_datadir/pdb_seqres/pdb_seqres.txt}
# make sure they all exist
for e in $( /usr/bin/env | grep "$AF_datadir" | cut -d= -f2 ) ; do
if [[ ! -e $e ]]; then
echo "missing $e -- check your environment "
exit
fi
done
export AF_preset="${AF_preset:-monomer}"
export AF_dbs="--uniref90_database_path=$AF_uniref90 --bfd_database_path=$AF_bfd --template_mmcif_dir=$AF_mmcif"
export AF_dbs="$AF_dbs --obsolete_pdbs_path=$AF_obsolete --pdb70_database_path=$AF_pdb70 --mgnify_database_path=$AF_mgnify"
if [[ $AF_preset =~ monomer ]]; then
export AF_dbs="--uniref90_database_path=$AF_uniref90 --bfd_database_path=$AF_bfd --template_mmcif_dir=$AF_mmcif"
export AF_dbs="$AF_dbs --obsolete_pdbs_path=$AF_obsolete --pdb70_database_path=$AF_pdb70 --mgnify_database_path=$AF_mgnify"
export AF_dbs="$AF_dbs --uniclust30_database_path=$AF_uniclust30"
else
export AF_dbs="--uniref90_database_path=$AF_uniref90 --bfd_database_path=$AF_bfd --template_mmcif_dir=$AF_mmcif"
export AF_dbs="$AF_dbs --obsolete_pdbs_path=$AF_obsolete --mgnify_database_path=$AF_mgnify"
export AF_dbs="$AF_dbs --uniclust30_database_path=$AF_uniclust30 --uniprot_database_path=$AF_uniprot --pdb_seqres_database_path=$AF_pdbseqres"
fi
# user customizable setup
export AF_docker="${AF_docker:---gpus all -u `id -u`:`id -g` --userns=host --security-opt no-new-privileges}"
export AF_mount="${AF_mount:--v /tmp:/tmp -v $HOME:$HOME -v /beegfs/desy:/beegfs/desy -v /beegfs/cssb:/beegfs/cssb -v /software:/software}"
export AF_image="${AF_image:-gitlab.desy.de:5555/frank.schluenzen/alphafold:2.1.1}"
export AF_outdir="${AF_outdir:-/tmp/alphafold}"
export AF_models="${AF_models:-model_1,model_2,model_3,model_4,model_5}"
export AF_env="${AF_env:---env TF_FORCE_UNIFIED_MEMORY=1 --env XLA_PYTHON_CLIENT_MEM_FRACTION=4.0 --env XLA_PYTHON_CLIENT_ALLOCATOR=platform}"
cat <<EOF
AlphaFold Setup
----------------------------------------------------------------------------------------------------
AF_datadir.: $AF_datadir
AF_docker..: $AF_docker
AF_mount...: $AF_mount
AF_image...: $AF_image
AF_outdir,,: $AF_outdir
AF_models..: $AF_models
AF_preset..: $AF_preset
AF_env.....: $AF_env
Hardware Setup
----------------------------------------------------------------------------------------------------
Host.......: $(hostname)
CPU........: $(grep "model name" /proc/cpuinfo | head -1 | cut -d: -f2 | grep -o '[a-Z].*')
GPU........: $(nvidia-smi -L |cut -d'(' -f1 | tr '\n' ' ')
Cores......: $(nproc)
Memory.....: $(free -g | grep Mem | awk '{print $2}')
Time.......: $(date)
Execute:
----------------------------------------------------------------------------------------------------
docker run $AF_docker $AF_mount --env LD_LIBRARY_PATH=$AF_NVL $AF_env $AF_image \
--output_dir=$AF_outdir --data_dir=$AF_datadir --model_preset=$AF_preset $AF_dbs "$@"
EOF
docker run $AF_docker $AF_mount --env LD_LIBRARY_PATH=$AF_NVL $AF_env $AF_image \
--output_dir=$AF_outdir --data_dir=$AF_datadir --model_preset=$AF_preset $AF_dbs "$@"
output
Messages like ....
/sbin/ldconfig.real: Can't create temporary cache file /etc/ld.so.cache~: Permission denied I1129 11:06:20.353542 140709785240512 templates.py:857] Using precomputed obsolete pdbs /beegfs/desy/group/it/ReferenceData/alphafold/pdb_mmcif/obsolete.dat. I1129 11:06:21.219282 140709785240512 xla_bridge.py:231] Unable to initialize backend 'tpu_driver': Not found: Unable to find driver in registry given worker: I1129 11:06:21.684458 140709785240512 xla_bridge.py:231] Unable to initialize backend 'tpu': Invalid argument: TpuPlatform is not available.
.... can be safely ignored.
databases
The databases to be used differ for monomers and multimers. The sample script (/software/alphafold/2.1.1/alphafold.sh) takes that into account. Trying to use all databases will result in an error (alphafold is unnecessarily picky), after initiating the docker container, which takes a few minutes.
multimers
Note: the fasta-file has to contain each chain as separate entry even if all sequences are identical. For the 1WUF sample it looks like this:
>1WUF_1|Chains A|hypothetical protein lin2664|Listeria innocua (272626) GHHHHHHHHHHGLVPRGSHMYFQKARLIHAELPLLAPFKTSYGELKSKDFYIIELINEEGIHGYGELEAFPLPDYTEETLSSAILIIKEQLLPLLAQRKIRKPEEIQELFSWIQGNEMAKAAVELAVWDAFAKMEKRSLAKMIGATKESIKVGVSIGLQQNVETLLQLVNQYVDQGYERVKLKIAPNKDIQFVEAVRKSFPKLSLMADANSAYNREDFLLLKELDQYDLEMIEQPFGTKDFVDHAWLQKQLKTRICLDENIRSVKDVEQAHSIGSCRAINLKLARVGGMSSALKIAEYCALNEILVWCGGMLEAGVGRAHNIALAARNEFVFPGDISASNRFFAEDIVTPAFELNQGRLKVPTNEGIGVTLDLKVLKKYTKSTEEILLNKGWS >1WUF_2|Chains B|hypothetical protein lin2664|Listeria innocua (272626) GHHHHHHHHHHGLVPRGSHMYFQKARLIHAELPLLAPFKTSYGELKSKDFYIIELINEEGIHGYGELEAFPLPDYTEETLSSAILIIKEQLLPLLAQRKIRKPEEIQELFSWIQGNEMAKAAVELAVWDAFAKMEKRSLAKMIGATKESIKVGVSIGLQQNVETLLQLVNQYVDQGYERVKLKIAPNKDIQFVEAVRKSFPKLSLMADANSAYNREDFLLLKELDQYDLEMIEQPFGTKDFVDHAWLQKQLKTRICLDENIRSVKDVEQAHSIGSCRAINLKLARVGGMSSALKIAEYCALNEILVWCGGMLEAGVGRAHNIALAARNEFVFPGDISASNRFFAEDIVTPAFELNQGRLKVPTNEGIGVTLDLKVLKKYTKSTEEILLNKGWS
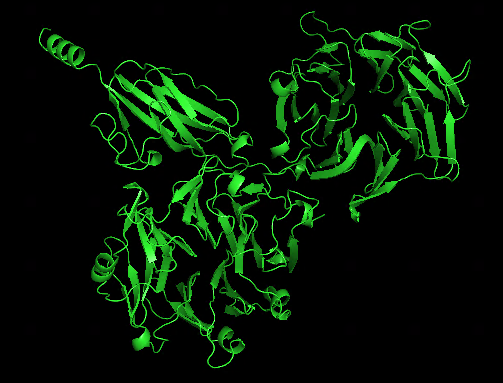
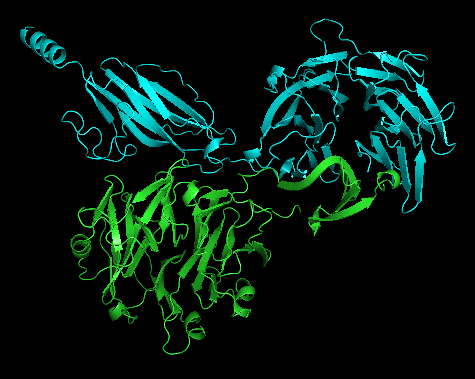
Just made some simple attempts using 1WUF.fasta as a template:
|
|
Original dimer structure 1WUF.pdb |
alphafold predicted dimer |

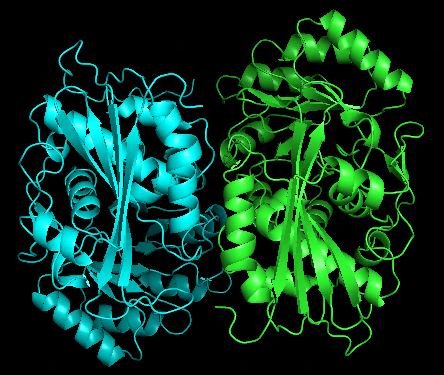
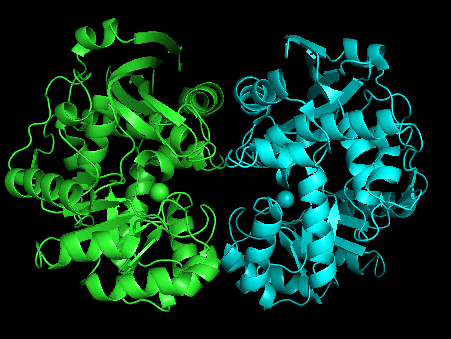

Another test run splitting T1050 into 2 domains and treat them as a multimer looks quite impressive:
alphafold prediction for T1050 |
prediction for 2 independent domains |