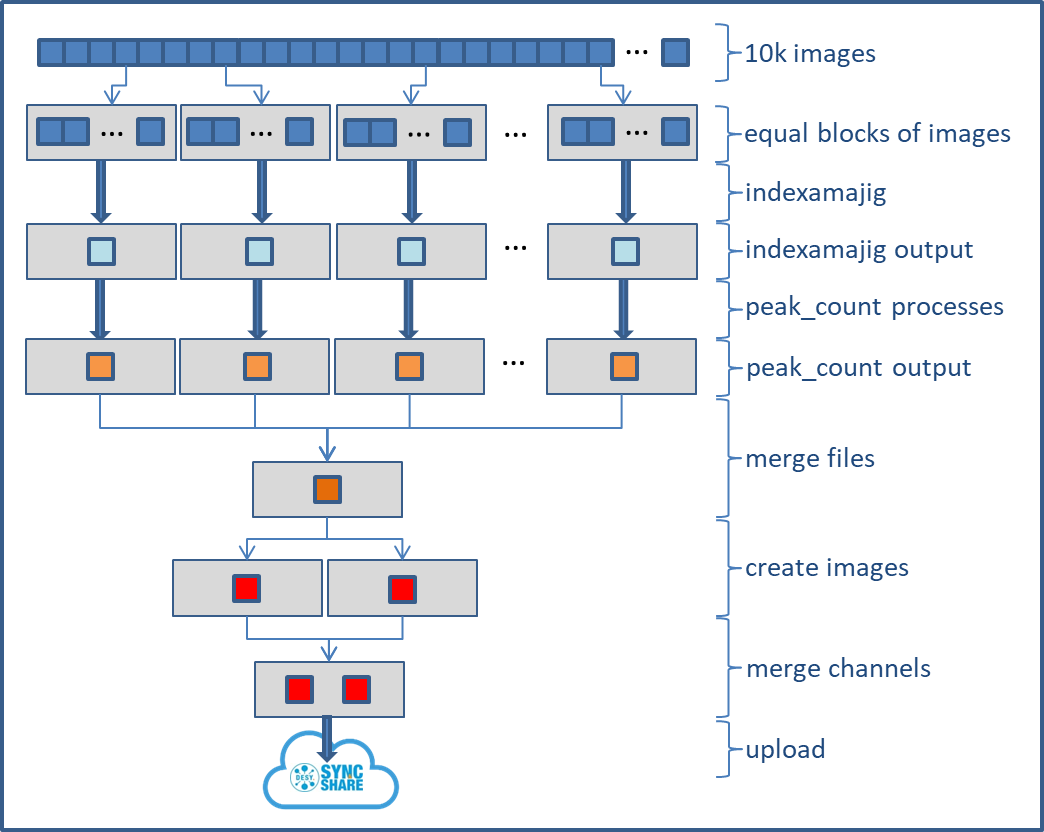
Tp provide a primitive example for swift data processing, I used crystfel offline processing in it's most simple form. For this particular case, I selected roughly 10k images from cxidb, split the set of images into bunches of roughly equal size, and distribute the jobs as slurm jobs on the cluster. swift takes care of the orchestration of the job submission, so you don't have to work with complicated constructs of job-arrays and dependencies.
The sample aims to illustrate very few of swift features. The image gives a rough idea of the workflow.
Content
The job submission script
A sample submission script could look like this:
In detail
The first part of the submission script just sets the environment. Please note that the "base job" allocates only a single node!
#!/bin/bash #SBATCH --time=0-01:00:00 # you need just a single node, kind of a master which orchestrates the jobs #SBATCH --nodes=1 #SBATCH --partition=upex #SBATCH --job-name=swift-split #SBATCH --dependency=singleton # I've chosen a name which reflects the number of nodes (58) to use #SBATCH --output=swift-split-58.out #SBATCH --tasks-per-node=1 # that's just to ensure that all jobs run on identical nodes for benchmarking #SBATCH --constraint=Gold-6240 # the basic setup export LD_PRELOAD="" source /etc/profile.d/modules.sh module load maxwell swift
The rest is the preparation of the input file, and running the actual workflow:
# it's easier to embed individual tasks into small scripts, and it's easier to have those in the PATH basedir=/beegfs/desy/user/schluenz/Crystfel.Swift export PATH=$basedir:$PATH # pass constraints and limits to slurm. It's also possible to declare those in the site config: export SBATCH_CONSTRAINT=Gold-6240 export SBATCH_TIMELIMIT=02:00:00 # use 58 nodes, so split the list of images into 58 roughly equal parts numnodes=58 mkdir -p $basedir/procdir # collect a list of images and count them ls -1 /beegfs/desy/group/it/ReferenceData/cxidb/ID-21/cxidb-21-run01[34]*/data1/LCLS*.h5 > $basedir/procdir/files.to.process num_images=$(ls -1 /beegfs/desy/group/it/ReferenceData/cxidb/ID-21/cxidb-21-run01[34]*/data1/LCLS*.h5 | wc -l) # split the files splitlines=$(( $(cat $basedir/procdir/files.to.process | wc -l) / $numnodes )) /usr/bin/split -l $splitlines -d $basedir/procdir/files.to.process $basedir/procdir/xxx # run the workflow swift -sites upex -config maxwell.conf indexamajig.swift -nsplit=$numnodes
type file;
# this generates just a list of images for each of the 58 parts and puts them into new set of files. There are simpler ways of doing that...
# splitter is the name of the scriplet. Note: you can't have '.' in your filename!
app (file o) split (int i, int nsplit)
{
splitter i nsplit stdout=filename(o);
}
# actually run indexamajig.
app (file o) indexamajig (int i)
{
indexamajigwrap i stdout=filename(o);
}
# collect information about number of peaks per image
app (file o) peak_count (file infile)
{
peak_count filename(infile) stdout=filename(o);
}
# ... and create a plot for peak counts
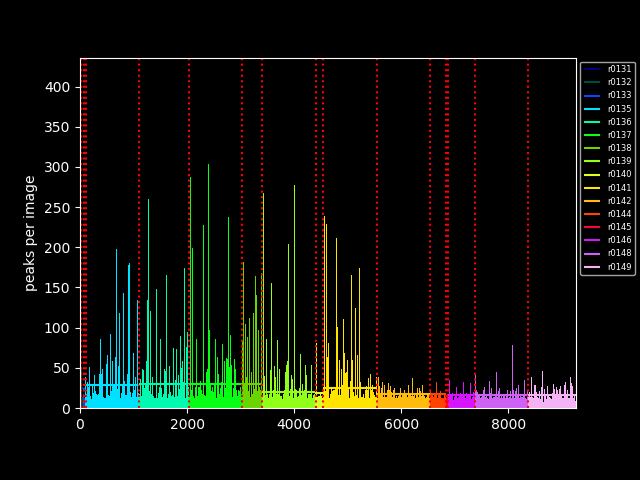
app (file o, file image) plot_peaks (file infile)
{
plot_peaks filename(infile) filename(image) stdout=filename(o);
}
# ... and cell dimension
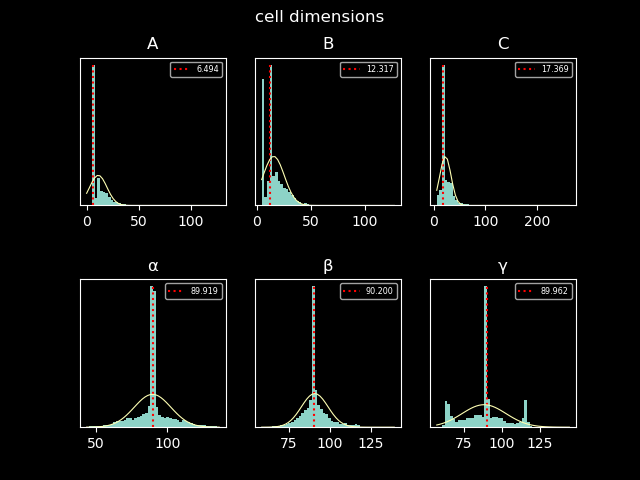
app (file o, file image) plot_cell (file infile)
{
plot_cell filename(infile) filename(image) stdout=filename(o);
}
# merge all peak information into a single file
app (file o) merge_peaks (file s[])
{
cat filenames(s) stdout=filename(o);
}
# if nsplit is not declared on the command line default to 16 nodes
int nsplit = toInt(arg("nsplit","16"));
file image_list[];
file peaks[];
foreach i in [0:nsplit-1] {
file splitout <single_file_mapper; file=strcat("output/split_",i,".out")>;
file indexamajigout <single_file_mapper; file=strcat("output/indexamajig_",i,".out")>;
file peak_countout <single_file_mapper; file=strcat("output/peak_count_",i,".out")>;
# create the lists of images
splitout = split(i, nsplit);
image_list[i] = splitout;
# process all sets of images. Though it looks serial it's actually done in parallel
indexamajigout = indexamajig(i);
# get number of peaks from indexamajib processing. With indexamajigout being the output of indexamajig and input for peak_counting, swift will schedule the job only when indexamajig is done.
peak_countout = peak_count(indexamajigout);
peaks[i] = peak_countout;
}
# combine the peak counting stats into a single file
file all_peaks <single_file_mapper; file=strcat("output/peaks.out")>;
all_peaks = merge_peaks(peaks);
# create two plots. again that's actually processed in parallel.
file cimage <"/beegfs/desy/user/schluenz/Crystfel.Swift/output/cells.png">;
file pimage <"/beegfs/desy/user/schluenz/Crystfel.Swift/output/peaks.png">;
file pp <single_file_mapper; file=strcat("output/pp.out")>;
file pc <single_file_mapper; file=strcat("output/pc.out")>;
(pc,cimage)=plot_cell(all_peaks);
(pp,pimage)=plot_peaks(all_peaks);
Results
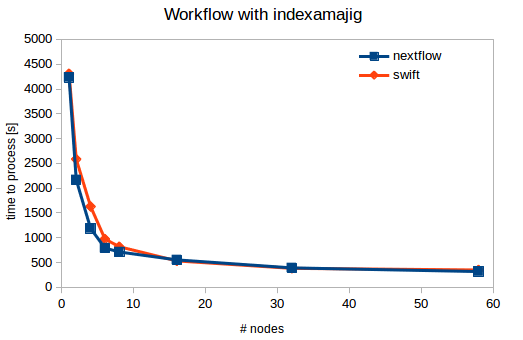
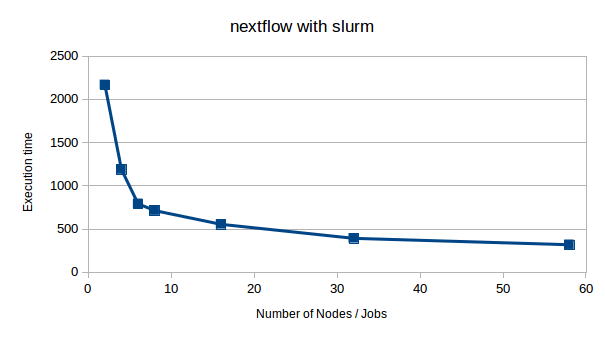
The scriplet above where used to run simple benchmarks. Execution of the code was done for various number of nodes in a single run, ranging from 1 to 58 concurrent jobs and nodes. Scaling is far from linear, but always also quite a bit of concurrent i/o which is presumably the limiting factor. The time to process the 9280 images can be reduced to about 300s, or 0.03s per image. Peak counting and plotting don't contribute significantly to execution times.
Just for sake of completeness, the two plot generated as part of the workflow:
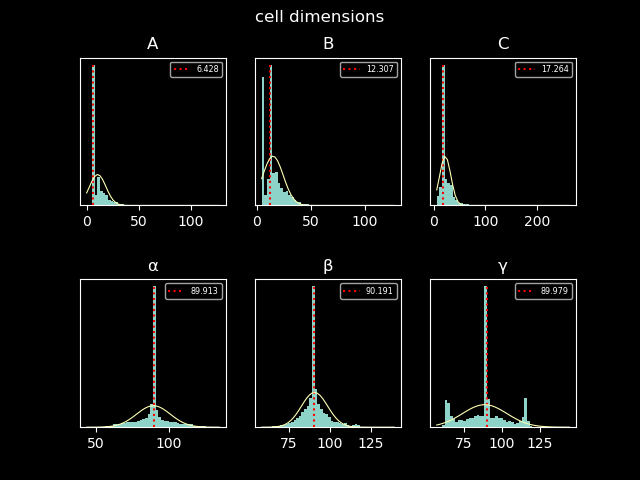
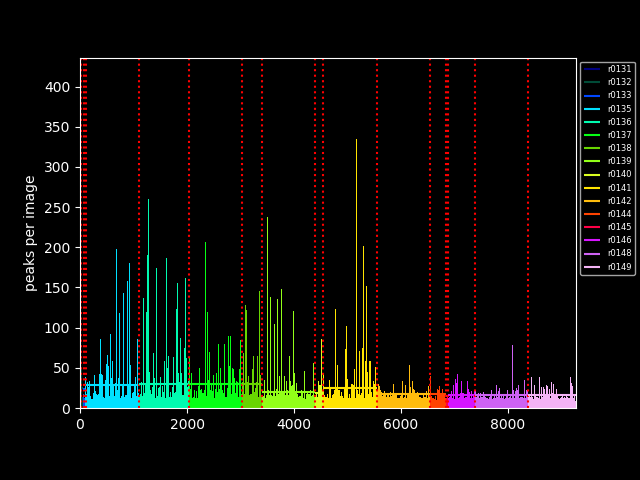
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}